

Sickle Cell Disease (SCD) is a genetic disorder characterized by the presence of mutated Hemoglobin S (HbS). It is an important but largely neglected risk to child survival in most African countries.
EPIDEMIOLOGY
It is common throughout much of sub-Saharan Africa, affecting up to 3% of births in some parts of the continent.
The most common subtype of SCD worldwide is homozygous SCD which is variably referred to as sickle cell anemia or Hb SS.
Sickle Cell Disease is responsible for significant illnesses and deaths, particularly amongst African and those from the Mediterranean.
The second subtype of SCD common in Africa is HbSC.
HOW DO THE ASSOCIATED ILLNESSES COME ABOUT?
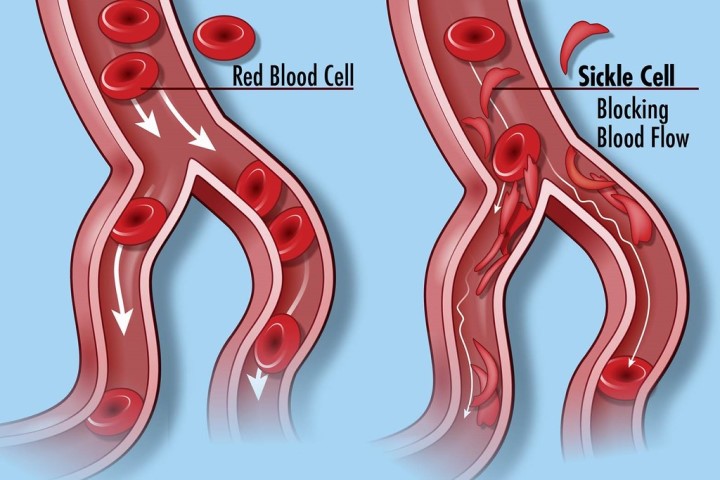
Normal red blood cells (RBCs) are spherical in shape, and in them are proteins known as hemoglobin which binds to oxygen and carries it to all parts of the body via the blood vessels. In someone who has SCD, the hemoglobin is abnormal, causing the RBC to assume a ‘sickle’-shape under certain conditions. These sickle-shaped RBCs have a shorter life span – compared to normal red blood cells that can survive for up to 120 days- thus resulting in the near-constant anemia that is characteristic in patients with this disease. In addition, because of their abnormal shapes, they tend to stick together more, blocking normal blood flow through the vessels, thus resulting in pain and other health problems such as stroke. The HbS also makes them prone to infections.
HOW DO PEOPLE INHERIT SICKLE CELL DISEASE?
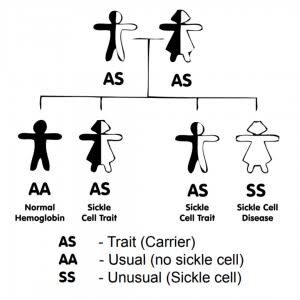
The commonest genotype of sickle cell disease is HbSS, and it is commonly referred to as sickle cell anemia.
Sickle cell disease is transmitted in an autosomal recessive mode of inheritance.
– If a ‘carrier’ father (AS) and a ‘carrier’ mother (AS) have a child, there’s a:
– 25% (1-in-4) chance of him/her having the AA genotype
– 50% (1-in-2 or 2-in-4) chance of him/her having the AS genotype
– 25% (1-in-4) chance of him/her having the SS genotype
Another variation of the sickle cell disease is HbSC. In this instance, a carrier parent (HbAS) and a parent with an abnormal form of Hemoglobin called ‘C’ (HbAC) have a 25% chance of giving birth to an offspring with HbSC. People with this genotype tend to have a milder form of SCD, though retinopathies (eye problems) may be commoner in them.
CLINICAL FEATURES
- For the first 6 months of life, majority of infants are protected largely by elevated levels of fetal hemoglobin (Hb F), and they therefore tend to present with symptoms later in childhood.
- A common presentation is hand-and-foot syndrome which is a form of dactylitis presenting in early childhood. It is characterized by bilaterally painful hands/feet
- In acute chest syndrome, young children present with chest pain, fever, cough, tachypnea, leukocytosis, and pulmonary infiltrates in the upper lobes of the lungs; adults are usually afebrile, dyspneic with severe chest pain, with multilobar/lower lobe disease.
COMPLICATIONS OF SICKLE CELL DISEASE
Sickle cell disease is associated with many complications and the commonest are painful crises. The commonest type of crises in people living with SCD is vaso-occlusive (painful) crisis. It could be caused by infection, stress, exposure to cold, dehydration. In some cases, the cause could be idiopathic (unknown). Other forms of crisis seen among SCD patients are aplastic, sequestration and haemolytic (hyperhaemolyitic) crises.
- There could be severe pain in the bones (limbs or back), joints, chest or abdomen
- In small children, painful crises usually manifest as dactylitis (painful swelling of bones of hand or foot).
Other potential complications include stroke, osteomyelitis (bone infection), eye problems, kidney problems, acute chest syndrome, leg ulcers amongst several others.
DIAGNOSIS
SCD can be diagnosed with a simple blood test, which involves hemoglobin electrophoresis. It is also possible to diagnose sickle cell disease whilst the baby is still in the womb. It’s also possible to check if an IVF embryo has SS genotype before implanting it in the womb of its mother. This is known as pre-implantation genetic testing.
HOW TO LIVE WITH SICKLE CELL DISEASE
People living with SCD require all the love and support as it is a tough condition to live with.
Non-medical measures to prevent or drastically reduce the frequency of complications include:
– Staying hydrated by drinking plenty of water: It is recommended that these patients drink at least 3.5L of water per day.
– Avoid extremes of temperatures as much as possible: Too much heat can lead to sweating which can result in dehydration, and excessive cold can trigger painful crises
– Stay away from places/activities associated with low oxygen levels: These can include places of high altitudes such as mountain tops or excessive exercise.
– Wash hands properly, prepare food thoroughly – These are steps to take in order to prevent infections
– Regular visits to the ophthalmologist (eye doctor) to check for vision loss, as eye problems may be problems.
Medical measures that can be taken include:
– Vaccines: Quite a number of SCD patients lose their spleen very early in life, and thus lose the protection that the spleen offers, in terms of curbing infections. Common vaccines that can be given to this effect include the influenza and pneumococcal vaccines amongst others.
– Malaria prophylaxis: When people living with SCD get malaria, it tends to be severe. As such, it is recommended that they take daily prophylaxis of antimalarials e.g proguanil, to reduce this risk.
– Hydroxyurea: Hydroxyurea has lately become commoner amongst people living with SCD. It has been shown to reduce the frequency of crisis amongst them, as it increases the amount of foetal hemoglobin (HbF) which carries the most amount of oxygen in the blood.
IS THERE A CURE FOR SICKLE CELL DISEASE?
The only well-researched and documented cure for Sickle Cell Disease (SCD) is a bone marrow or stem cell transplant. This entails transferring compatible and healthy cells from the bone marrow of a donor to someone who has unhealthy bone marrow. This procedure – as with all surgeries – comes with its potential complications.
WAY FORWARD
Prevention is better than cure; reducing the incidence of birth of SCD sufferers remains the best bet. Genotype testing for intending couples would go a long way in achieving this. Giving support and love to people living with SCD goes a long way in making their lives easier, and taking measures to curb the frequency of illnesses helps them to live their best lives possible!


